Revised for Spring 1997
We have tried many different methods in Chem 102 in an attempt to give a varied experience to the student. Your current textbook may or may not cover the experiments chosen. The notes below contain methods, tips, tricks, and techniques in carrying out these methods. Not all of the notes will apply to the experiments chosen for this semester, so disregard any material that does not apply to the current laboratory schedule.
The Internet version of these lab notes may contain corrections or additional information.
The Lab Notebook
You must use a bound laboratory notebook. Reserve the first four pages for a table of contents, then number all succeeding pages. Leave the left hand page blank for scratch. Put all hard data and calculations on the right-hand side. All entries must be in ink. Nothing can be erased; pages cannot be removed. It is very important that all data be entered directly into your notebook. Do not use paper towels or other intermediate pieces of paper to record data for later transferal into your notebook. Leave ample room for data tables so that all of your data for a given run will fit into one place. If you look below at the suggested format for your lab notebook, you will find that there is provision for a Data Summary where you can make everything very neat.
Each experiment should follow this format:
1. Title
2. A brief theoretical discussion of the chemistry involved. Include all relevant chemical reactions and mathematical procedures. Do this before you start the experiment. The purpose is to get you to organize all of the theoretical details so they are clear in your mind. Don't skip this step just because the first few experiments are so straightforward. Get used to doing this now, because later we will be doing some very messy things that will require organization beyond what is given in your text.
3. A skeleton outline of the experimental procedures used in the experiment. Only rarely will we be doing the experiment exactly as specified in the book or on a handout. This outline, done before you start the experiment, will allow you to collect all relevant details into one place. Do not rely on a handout to tell you the corrections and additions. It is expected that students in this course will demonstrate the ability to do independent organization of a project.
4. Data Tables -- make these as clear and as neat as possible. Be sure all data are properly labeled so you can find it later. Carry your notebook to the balance or other instrument for direct entry of all data. If you find things get a little messy at this point, don't become overly concerned. Just be sure you can identify all numbers two weeks later. Notebook grades are given on clarity, not superficial neatness.
5. Data Summary -- here is your big chance to be neat. Gather together all of the data that is required to perform the final calculations. Place this data into nice neat data tables. You may wish to look at journals such as Analytical Chemistry to see how data tables are set up by the professionals. With your data all in one place, performing the calculations will be made much easier. If disaster strikes, and you make a calculation error, your data tables will make finding the error much easier. You can also compare these data with the originals in the same book.
6. Computation of Results -- Have at least one annotated sample calculation complete with units and values for all constants, such as gram formula weights. Graphs and other computer outputs should be formatted for cutting and pasting into your notebook. If cutting and pasting of computer output is not feasible, label the output clearly and keep it in a separate folder. Computers can easily generate reams of graphs and tables. Include only what is required for your experiment. Points may be deducted for irrelevant junk.
7. Statistical analysis of results -- Use of 95% confidence limits, linear least squares, or other techniques is required. All reported values must include a calculated error. We will be making routine use of computer spreadsheets, statistical packages, and graphics programs in this course. Remember that the purpose of all this computing power is to generate accurate results and appropriate error analyses, not to snow your readers with irrelevant output.
8. Conclusions -- Most experiments conclude with a final reported result, expressed to the correct number of significant figures, and with a 95% confidence limit. You will be graded on this result. Follow this number with a written statement explaining the reliability of this number. Comment on the sources of error and make suggestions on how future experimenters might make improvements. Be sure you make use of your statistical analysis in this part of the notebook report.
You will never surrender your notebook to me for grading. We will go over it together at the time you report your results. This will go very quickly if your notebook is well organized. Grades will be awarded on the spot.
Other Required Materials
You are required to wear Safety Glasses in the laboratory at all times. Purchase a pair that is comfortable and has good optical quality. See the lab safety handout for more details, including precautions for contact lens wearers.
Be sure your calculator has a full range of scientific and statistical functions. We do a lot of calculations in this course. Titration curves and other larger data sets will be done using the Macintosh computers available in S 232 and elsewhere on campus. You may also use your own computer spreadsheet if you have one. Some computers, such as the five of the ten in S 232 that are not yet upgraded with hard drives, require Startup Disks. These are available at the Kennel Bookstore. You will need about three 3.5 inch, double-sided double density computer disks to use these. Warning: Eight of the ten computers in S-232 are old Macintoshes that can only use the 800K double-sided, double-density (2DD) disks. Do NOT use the newer High Density (HD) disks on these old machines. Their disk drives will not magnetize the newer media with any reliability. HD disks seem to work at first, but then fail just when you are trying to finish your report. I have seen this happen.
Camera stores sell white cotton gloves used for handling color print materials. Some cosmetic counters carry them also for applying makeup. These work very well to keep fingerprints off glassware in the balance room. Purchase of these is optional, but you will find them a great convenience.
About Computers in the Course
We will be making extensive use of the Microsoft Excel Spreadsheet on the Macintosh computers for working up lab data and solving other problems in the course. You will need a Student Startup Disk and some data disks if you want to use the five computers in S-232 that have not been upgraded. These are available at the bookstore. You may also use your own computer if you have a spreadsheet capable of doing true X-Y scatter plots as well as the usual calculations. If you have been considering buying your own computer, now would be a good time to do it. Substantial student discounts are available on computers and software, such as Microsoft Office (either PC or Macintosh) which includes both a word processor and the spreadsheet (Excel) that you will need. All students will be given an e-mail account on the campus student mail server. Some course materials and announcements will be distributed only by e-mail or by HTML documents accessed on the Internet via lynx or Netscape. Use of this account will be required. If you have a computer at home, you will need a modem to contact the campus over the telephone lines. The Macs in S-232 are wired into the campus network and can reach the student e-mail server via MacIP. The five machines that are not upgraded will require a modified startup disk available from your instructor. (Bring him a blank 2DD disk.) Netscape only runs on the five upgraded machines.
Notes on the Experiments:
1. Check in and balance instruction -- This will be covered in the lab. Your introduction to the balance and many of the concepts involved should be considered part of the lecture material. Choose a clean and dry weighing bottle and weigh this bottle on all ten electronic balances. Enter your masses on the posted form provided. The data collected will be statistically analyzed and used to determine if there are any problems with the balances or with the student's ability to use them.
2. Gravimetric Analysis of Chloride -- You will be issued enough Chloride Unknown to do both this experiment and the next one on Volumetric analysis. You will also be given a bag of silver nitrate that is used to prepare 1000 mL of 0.1M AgNO3 -- this too must last through both experiments.
Clean and dry all of your weighing bottles. Put an identifying mark on each bottle with a graphite pencil on the frosted areas provided. Do NOT use paper labels. Weigh each one (cover on) and record this TARE weight in your laboratory notebook. Transfer your chloride unknown to one or more weighing bottles (never fill over two-thirds full); do the same with the silver nitrate. Dry both materials for one hour at 110deg.C. Weigh again to find out how much material you have been given. Weigh all samples by difference from these bottles. The technique will be demonstrated in lab.
See Drying and Weighing for a description of these techniques. Be sure and check the Drying and Weighing page for directions in making up the silver nitrate solution in the next step.
Weigh out enough silver nitrate to make up the 0.1M solution. Remember, the weight need be only within ten percent of the nominal, or TARGET value required.
Clean and dry three medium or fine-porosity sintered glass crucibles. The cleaning procedure may involve the use of concentrated nitric acid. Be very careful not to spill nitric acid on your skin. Bring these crucibles to constant weight with repeated dryings.
Additional procedures are in the book or are found in the excerpt given below:
Gravimetric Determination of Chloride
from Skoog, West & Holler, Fundamentals of Analytical Chemistry, 6th Edition, Saunders, 1992
Procedure
Clean three sintered-glass or porcelain filtering crucibles by allowing about 5 mL of concentrated HNO3 to stand in each for about 5 min. Use a vacuum (Figure 33-17) to draw the acid through the crucible. Rinse each crucible with three portions of tap water, and then discontinue the vacuum. Next, add about 5 mL of 6 M NH3 and wait for about 5 min before drawing it through the filter. Finally, rinse each crucible with six to eight portions of distilled or deionized water. Provide each crucible with an identifying mark. Bring the crucibles to constant weight by heating at 110deg.C while the other steps in the analysis are being carried out. The first drying should be for at least 1 hr: subsequent heating periods can be somewhat shorter (30 to 40 min).
Transfer the unknown to a weighing bottle and dry it at 110deg.C (Figure 33-9) for 1 to 2 hr; allow the bottle and contents to cool to room temperature in a desiccator. Weigh (to the nearest 0.1 mg) individual samples by difference (page 5324) into 400-mL beakers (Note 1). Dissolve each sample in about 100 mL of distilled water to which 2 to 3 mL of 6 M HNO3 has been added.
Slowly, and with good stirring, add 0.2 M AgNO3 to each of the cold sample solutions until AgCl is observed to coagulate (Notes 2,3); then introduce an additional 3 to 5 mL. Heat almost to boiling, and digest the solids for about 10 min. Add a few drops of AgNO3 to confirm that precipitation is complete. If additional precipitate forms, add about 3 mL of AgNO3, digest, and again test for completeness of precipitation. Pour any unused AgNO3 into a waste container (NOT into the original reagent bottle). Cover each beaker, and store in a dark place for at least 2 hr and preferable until the next laboratory period.
Read the instructions for filtration in Section 33G-2. Decant the supernatant liquids through weighed filtering crucibles. Wash the precipitates several times (while they are still in the beaker) with a wash solution consisting of 2 to 5 mL of 6 M HNO3 per liter of distilled water; decant these washings through the filters. Quantitatively transfer the AgCl from the beakers to the individual crucibles with fine streams of wash solution; use rubber policemen to dislodge any particles that adhere to the walls of the beakers. Continue washing until the filtrates are essentially free of Ag+ ion (Note 4).
Dry the precipitate at 110deg.C for at least 1 hr. Store the crucibles in a desiccator while they cool. Determine the weight of the crucibles and their contents. Repeat the cycle of heating, cooling and weighing until consecutive weighings agree to within 0.2 mg. Calculate the percentage of Cl- in the sample.
Upon completion of the analysis, remove the precipitates by gently tapping the crucibles over a piece of glazed paper. Transfer the collected AgCl to a container for silver wastes. Remove the last traces of AgCl by filling the crucibles with 6 M NH3 and allowing them to stand.
Notes
1. Consult with the instructor concerning an appropriate sample size.
2. Determine the approximate amount of AgNO3 needed by calculating the volume that would be required if the unknown were pure NaCl.
3. Use a separate stirring rod for each sample and leave it in its beaker throughout the determination.
4. To test the washings for Ag+, collect a small volume in a test tube and add a few drops of HCl. Washing is judged complete when little or no turbidity develops.
3. Glassware Calibration -- although a day has been put into the schedule for this, it is best done while drying crucibles during the gravimetric chloride experiment. We rarely find poorly calibrated glassware, but the techniques required will test both your ability to weigh and to dispense liquids accurately. Calibrate two of your pipets (the 10 mL and the 25 mL) and your buret. Do each pipet three times and calculate the Confidence Limit for your measurements. Compare this measure of precision with the accuracy (your value - stated value) shown by your measurements.
In the case of the buret, record the volume and the mass dispensed every five mL by doing successive weighings of the water dispensed into your glass stoppered flask. In a table in your notebook, record the volume dispensed to the nearest 0.01 mL, and the mass of water. Using a computer spreadsheet, compute the volume from the mass and subtract it from the volume as read from the buret. This will be the absolute error, in mL, at each 5 mL mark on the buret. Finally, plot the error vs. volume dispensed to make an error chart for your buret. Print the spreadsheet out and paste it into your laboratory notebook.
Have a brief conclusions section where you summarize your data and errors, and comment on the quality of your glassware.
4. Volumetric Chloride (revised) -- this is done on the same sample used for the grav analysis. The first part of the experiment will be a simple determination of %Cl- in a solid unknown using either the Mohr Method, or the Fajans adsorption indicator method as directed. See the procedures below, as well as the material in your text book.
As an additional experiment we will add a mixed halide potentiometric end point using a silver wire sensing electrode and a double junction reference electrode. (Alternatively, we may try the method described in Harris, where a glass electrode and silver electrode are used, but results from this method have been poor in the past.) Several data points need to be taken both before and after each end point for the Grans Analysis; good data need to be taken during the end point transitions for the "eyeball" method of simply estimating the end point from the breaks in the titration curve.
The potentiometric end point is a separate experiment, reported in a brief lab report consisting of (1) spreadsheets of your data and Grans calculations, (2) computer-generated graphs of the titration curve and of the Grans Plots, and (3) an estimate and discussion of the error in the end points found. When reporting your results, be sure to summarize your findings in a table to ensure that nothing is forgotten. In this experiment we have two end points (one for chloride and one for iodide) and three methods for estimating each end point ("eyeball" the unmodified titration curve, front Grans Plot, back Grans plot). In this table the error estimates are to be based on how much scatter is inherent in the method used, and not on any comparison with actual millimoles in the sample. Your graphs should show how you did your error estimates.
Iodide End Point
in mL
|
Chloride
End Point
in mL
| |
| "eyeball"
titration curve
|
Value
+/- Error
|
Value
+/- Error
|
| Front
Grans
|
Value
+/- Error
|
Value
+/- Error
|
| Back
Grans
|
Value
+/- Error
|
Value
+/- Error
|
Any comparison with actual millimoles present should be done in the Discussion, where you can talk about whether or not your value is significantly different from the true value (if you have a true value you can use).
Prepare your own "unknown" by adding 1.0 millimoles of KI to 2.0 millimoles of your primary standard NaCl (measure the mass of the NaCl very accurately). Compare the amount of NaCl found by the Gran's Plot end point done on a spreadsheet with the amount of NaCl that you added. (Calculate the KI too, but we cannot do an accurate millimole comparison on the KI, since it is not a primary standard.) Since the molarity of the AgNO3 is based on the Mohr or Fajans titration, you can compare how well the Potentiometric Method agrees with the Indicator Method. The Grading Sheet used for this report is available from your instructor.
If your text does not contain the Mohr Method and your instructor elects to use it, here it is (modified from Skoog, West & Holler, Fundamentals of Analytical Chemistry, 6th Edition.)
You should still have 0.1 M AgNO3 solution left over from the Gravimetric Method. Also prepare 5% Potassium Chromate by dissolving 1.0 g of K2CrO4 in 20 mL of distilled water. The storeroom may have already prepared this solution. You must standardize the AgNO3 solution against primary standard NaCl that has been dried for 1 hr. at 100deg.C and desiccated at least overnight. Titrate at least three replicates (or until you have good statistics) using the Mohr Method.
Procedure: Dry the unknown at 110deg.C for at least 1 hr. Cool in a desiccator overnight. Compute your sample size based on a 30 mL titration, using the %Cl computed from the gravimetric determination. Place each sample in a 250 mL conical flask, and dissolve in 100 mL of distilled water. Add a small quantity of NaHCO3 to make the solution basic, then add about 2 mL of the 5% potassium chromate solution. Titrate to the first permanent appearance of red Ag2CrO4.
The Fajans Method. A more environmentally acceptable end point avoids the use of chromate as an indicator, using the adsorption of 2'-7'-dichlorofluorescein on the surface of the AgCl precipitate. Prior to the equivalence point the AgCl has a net negative charge due to excess Cl-, repelling the indicator dye. When the solution has excess Ag+, however, the adsorbed positive charge attracts the dye, giving a bright pink color. The procedure below is from Skoog & West, Fundamentals of Analytical Chemistry, 4th Edition.
The storeroom will prepare the indicator by dissolving 0.1 g of dichlorofluorescein in 100 mL of 75% (v/v) ethanol/water solution.
Method 2-1. Determination of Chloride by the Fajans Method
Reagents needed:
Standard AgNO3 solution.
Dichlorofluorescein solution
Dextrin
Procedure. Dry the unknown at 110deg.C for 1 hr. Weigh samples into conical flasks and dissolve in an appropriate volume of distilled (or deionized) water (Note 1). Add about 0.1 g of dextrin and 5 drops of indicator. Titrate (Note 2) with AgNO3 to the first permanent appearance of the pink color of the indicator.
Notes
1. Weigh (to the nearest 0.1 mg) 0.25-g samples if 0.1 M AgNO3 is to be used and about half as much for the 0.05 M reagent. Use about 200 mL of water for the former and 100 mL for the latter. For 0.01 M AgNO3, it is recommended that a 0.4-g sample be weighed (to the nearest 0.1 mg) into a liter volumetric flask and that 50.0-mL aliquots be taken for titration.
2. Silver chloride is particularly sensitive to photodecomposition in the presence of the indicator: the titration will fail if attempted in direct sunlight. Where this problem exists, the approximate equivalence point should first be ascertained by a trial titration, this value being used to calculate the volume of AgNO3 required for the other samples. The addition of indicator and dextrin should be delayed until the bulk of the silver nitrate has been added to subsequent samples, after which the titration should be completed without delay.
5. Sodium Carbonate (revised) -- We will base this on the method HCl titration in the book, followed by a complete titration curve done with a pH meter. An extensive analysis of the end points will be done using Microsoft Excel on the Macintosh computers. See the handout on the Carbonate Analysis for more details. Be sure to report your final results in a Table, similar to that described under the Volumetric Analysis of Chloride. This table will have more end point methods in it, of course. Primary emphasis will be on the end point detection methods. You can earn extra credit toward removing missed points on the report if you also compare your data to data simulated from your actual millimoles, but this is optional. The Grading Sheet used for this report is available from your instructor.
6. Complex Formation -- We will need to modify most textbook procedures to use solid unknowns. Follow the procedure below:
Chemistry 102 and 105
Revised Spring 1987
The procedures in your text are probably designed for aqueous samples containing calcium and magnesium. (See the references in your lab schedule to find the procedure for "hard water" analysis.) To use solid unknowns and standard materials, you will need to dissolve them first. Since calcium carbonate will not dissolve in distilled water, you will need to follow the directions below before completing the titrations as directed in your text. We have found that the purity of our EDTA reagents is not good enough to use it as a primary standard. Directions for preparing a standard calcium solution are also given. Titrate this solution in the same way you titrate your unknown solution; then calculate the molarity of the EDTA.
1. Your instructor will provide you with bags of unknown, of primary standard calcium carbonate, and of the EDTA. Dry the unknown and the calcium carbonate for one hour at 110deg. C. The EDTA should not be dried; just put it in a weighing bottle.
2. Prepare 500 mL of 0.01M EDTA. The salt is usually Na2H2Y.2H2O. (Y = EDTA) Compute the required mass, weigh it out, and make up to the mark with distilled water in a 500 mL volumetric flask. The exact molarity of this solution must be determined by standardizing against standard calcium carbonate.
3. Many EDTA procedures require that traces of magnesium be added to your solution. To avoid adding any additional titratable cation (Mg2+) we add the magnesium as MgY2-. To prepare this complex (hereafter referred to as "the spike"), weigh out 0.4 millimoles of Mg2+. Check the reagent bottle for composition and gram formula weight needed to calculate the mass. The salt is usually MgSO4, but could be something else. Dissolve in 50 mL of distilled water, and titrate according to the hard water instructions in your text. In order to experiment with the end point, you can add a crystal of Mg salt to restore the red color, then run through the end point again. Stop at your best estimate of the end point color. Store the solution in your glass stoppered Erlenmeyer flask. Label it "Mg Spike Solution." When titrating either standard calcium, or a calcium unknown, add about one mL of this spike to your solution in addition to the usual buffer and indicator.
4. Dissolution of solids containing calcium carbonate requires special procedures--this includes both your standard calcium carbonate as well as your unknown. Once the solid is weighed out (see below) transfer it to a 250 mL Erlenmeyer flask, and add about 100 mL of water. Add 6M HCl dropwise and swirl the flask to dissolve the calcium carbonate. Warm after each addition to expel the carbon dioxide. Continue until all of the solid has dissolved. (In the case of many unknowns, a small amount of insoluble white silica may remain.) Avoid a large excess of the HCl. After dissolution is complete, quantitatively transfer to a suitable volumetric flask and dilute to the mark. Shake for five to ten minutes to achieve uniformity.
5. To prepare a standard solution, weigh enough dried calcium carbonate standard to allow you to use 25 mL aliquots from a 250 mL volumetric flask. (The calculation of the mass is left as an exercise for the student. Remember that each titration will require at least 0.3 millimoles of calcium.) After preparing the solution and mixing it well, transfer to another container to free the volumetric flask for your unknown.
6. The unknown solution is prepared by dissolving about one gram of unknown (see step 4) and making up to 250 mL in a volumetric flask.
7. The percentage of CaO in the unknowns can vary considerably from student to student. Try a 10 mL aliquot first. If the titration volume is too small, choose an appropriate larger aliquot size.
8. Don't forget to report %CaO. This is the standard way to report rock and mineral analyses. It does not depend on the actual compound of calcium in the sample.
Note: Buffer and indicator solutions are available in the lab. Be sure you use the high buffer capacity pH 10 solution designed for this experiment. Do not confuse it with the low buffer capacity pH 10 solution used to calibrate pH meters. pH paper will be available to check the pH of your solutions if your end point seems too drawn out due to low pH. The indicator will be either Calmagite or Eriochrome Black T. See your instructor.
7. Spectrophotometric Iron (revised) -- Harris gives a procedure for determining Fe in water or vitamin tablets using o-phenanthroline. Skoog, West and Holler have an experiment on measuring Fe in a Natural Water. You will need to modify these experiments so they can be used on a solid ore sample. You will need to consult your text or other sources to find out how to dissolve your sample. You will have to find a source of Standard Iron to prepare standards. (Hint: Check out Ferrous Ammonium Sulfate.) Then you will need to devise a suitable dilution scheme to bring the concentrations down into the range appropriate for this method. Doing this accurately will require careful planning on your part. Remember that you will need to use the volumetric ware found in your desk to carry this out. Your final colored solutions should end up mostly in the 50 mL volumetric flasks provided as part of your standard kit. Volumes of less than 5 mL cannot be measured with sufficient accuracy, so serial dilution schemes will be required to allow use of your standard buret and pipets. This experiment closely follows the processes needed to adapt standard procedures in Real Life. Once you have figured this all out, you get to change all the concentrations so you can run the sample by atomic absorption as well.
Our storeroom will provide the o-phenanthroline, hydroxylamine hydrochloride, and sodium acetate solutions as used in the spectro iron procedure found in Skoog and West. Your text may differ, in which case get the Skoog and West procedure from your instructor.
8. Determination of Glucose -- See the procedure below. Make sure you wade through all of the chemistry and have a good outline in your laboratory notebook. It is easy to mess up this experiment if you don't know what you are doing.
Reducing sugar (glucose for example) determinations are routinely run on samples of blood, urine, various beverages and food products. A general method for determining reducing sugar follows.
It is common knowledge that Fehling's solution (which contains Cu2+) is reduced by glucose to form a brick-red precipitate of Cu2O. The amount of Cu2O formed may be determined by oxidizing it back to Cu2+, using free iodine. The iodine used for this purpose is generated in the solution by the reaction of KI with KIO3 in the presence of acid:
5 I- + IO3- + 6 H+ ---> 3 I2 + 3 H2O (1)
Oxidation of the Cu2O precipitate may be represented by equation (2)
Cu2O + I2 + 2 H+ ---> 2 Cu2+ + H2O + 2 I- (2)
Equation (1) forms a known quantity of excess iodine. After reaction (2) is complete, the remaining iodine (which did not react with the Cu2O) is determined by titration with standard sodium thiosulfate solution using starch indicator:
2 S2O32- + I2 ---> 2 I- + S4O62- (3)
The overall reaction that defines the equivalent weight of KIO3 is the following:
IO3- + 5I- + 6H+ + 6S2O32- ---> 6I- + 3S4O62- (4)
Where iodate to iodide is a six electron change.
Solution A: Dissolve 34.64 g CuSO4.5H2O in water and dilute to 500 ml.
Solution B: Dissolve 173 g sodium potassium tartrate and 50 g NaOH in water and dilute to 500 ml.
(A and B will be available in the laboratory.)
Standard potassium iodate solution: Weigh to the nearest 0.1 mg enough dry KIO3 (110o, 1 hr) to prepare 500 ml of about 0.1 N (1/60 M) solution. Quantitatively transfer the salt to a 500 ml volumetric flask and dissolve in a small amount of water. After first making the solution basic with 3-4 drops of 6 N (6 M) NaOH add about 20 g of iodate-free KI. Dilute to 500 ml.
Sodium thiosulfate solution: 0.12 N (0.12 M). Dissolve 19 g of Na2S2O3 (or enough Na2S2O3.5H2O to give 19 g of Na2S2O3) in 1 liter of distilled water. Add about 0.1 g of Na2CO3 to keep the solution neutral or slightly alkaline and thereby retard decomposition to elemental sulfur. If you wish, you may prepare this solution quantitatively as a check on the normalities (molarities). See Skoog, West and Holler, Fundamentals of Analytical Chemistry, 7th Ed., p. 363, or your current text, for additional precautions regarding thiosulfate solutions.
Starch indicator (Make daily): Suspend 1 g of soluble starch in 100 ml distilled water. Bring to a boil. Cool and use 1-2 ml per titration.
Saturated potassium oxalate: Available in laboratory.
Dry the sugar sample for not more than 1 hour at 95oC. Weigh about 2 grams of the sample to +or- 0.1 mg and place it in a 500 ml volumetric flask. Dissolve in about 300 ml of water and then fill to the mark. Mix thoroughly. Prepare a boiling water bath of 1 liter of distilled water in a 2-liter beaker. Rapidly pipette 25 ml each of Fehling's solutions A and B into a 500 ml Erlenmeyer flask. Complete the following analysis before doing additional runs. Add exactly 50 ml of the glucose solution prepared above. (Note: If a sample smaller than 50 ml is to be analyzed, the total volume should be brought to a100 ml at this point by addition of water). Cover the flask with a small inverted beaker and place in the boiling water bath for exactly 10 minutes. (Note: The bath must be large enough to boil the entire 10 minutes.) After 10 minutes immediately cool the flask to room temperature with running water or ice bath.
Add 20 ml of saturated potassium oxalate solution and mix. This prevents future oxidation of iodide by copper (II) ions and dissolves any Cu2I2 formed. Pipette 50 ml of KIO3 solution into the flask followed by 16 ml of 6 N (3 M) H2SO4 (measure with a graduate). Shake the solution gently until the copper(I) oxide is completely dissolved. If insufficient iodine is evolved add another portion of KIO3 solution or start with a smaller aliquot of sugar sample.
Titrate the remaining I2 (actually brown I3-) with sodium thiosulfate solution, using starch indicator. The starch should be added just before the endpoint is reached, as indicated by the color of the solution. (Why?)
Standardize the sodium thiosulfate solution by repeating the above procedure, omitting the Fehling's solutions and potassium oxalate, substituting 120 ml of distilled water for the sugar sample and using 25 ml of KIO3 solution instead of 50 ml. Calculate the normality (molarity) of the sodium thiosulfate solution. Calculate the copper equivalent for your unknown as mg of copper in the Cu2O. Convert mg Cu to mg glucose using one of the methods suggested below and report your answer as mg glucose/100 ml based on an unknown sugar sample weighing exactly 2.000 grams.
Methods for converting mg Cu to mg glucose:
(1) If a chemistry handbook or other source of Munson-Walker-Hammond tables is available, read mg glucose equivalent to your mg Cu directly from the table.
(2) From an accurately plotted graph or an equation based on the following Munson-Walker data as determined by Hammond, determine the Cu-glucose weight ratio.
Cu in mg
|
Cu-glucose
ratio
|
| 400
|
1.863
|
| 350
|
1.892
|
| 300
|
1.921
|
| 250
|
1.950
|
| 200
|
1.978
|
| 150
|
2.008
|
| 100
|
2.041
|
|
70
|
2.059
|
|
50
|
2.075
|
Divide the amount of copper in your sample by the Cu-glucose ratio.
Both of these methods give glucose in mg/50 ml if a 50 ml sugar sample was used. Please remember that you are to report your answer as mg glucose/100 ml, based on a glucose sample of exactly 2.000 grams. Correct your result for the actual mass of sample used.
9. Ion Exchange, HPLC, GLC, or Fluoride by Ion Selective Electrode -- More details on the IE/HPLC/GLC experiment will be given in class. If you elect the Fluoride experiment, you provide your own sample of fluoridated toothpaste or tap water (the tap water must be known to have measurable fluoride). We will want to do this one both by Working Curve and by Standard Addition. Pay close attention to possible matrix effects. Get replicate results to verify precision. This experiment requires a full written report, which means an Abstract, a Theory Section, a Data Section, a full workup of Results, with graphs and error analysis, and finally a Conclusion that must show the reader why your results should be believed. See the handout "The Chemistry 106 Lab Report for 102" for details on what is expected in a report of this type.
Depending on availability of equipment, we may have to be creative on planning for this last experiment. Experiments carried out in the past include fluoride analysis, gas chromatography measurement of alcohol in beverages (you must be over 21 to do this experiment), and high performance liquid chromatographic determination of uric acid and other components in urine. To do this final project, you need to do two things:
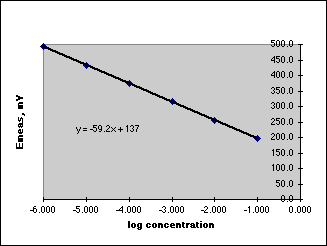
A. Begin with a basic experiment, usually from a textbook containing a student experiment. In the experiments done to date, the experiment from a textbook only gives directions for a Working Curve method for the unknown determination. The working curve for Fluoride, for example, might look like:
Using this working curve, you run your unknown, get a Response, and determine its concentration from the working curve. Repeat this a few times to get some statistics. Then work back through your dilution scheme to determine the concentration in your unknown. But having used this working curve, the experiment is only half done. The working curve is prepared from pure standards, but our unknown contains a matrix which may or may not affect the results. You will be unable to prove in your report that the value obtained for your unknown is reliable.
B. Using the methods of part A, devise a Standard Addition Method that will allow you to run the determination in the presence of the unknown matrix. In general, you measure your unknown, then add a known amount of analyte to it so that you get about a 40% increase in the Response, and measure it again. Repeat this two or more times to get something you can plot. In the case of Fluoride, the standard addition plot looks like:
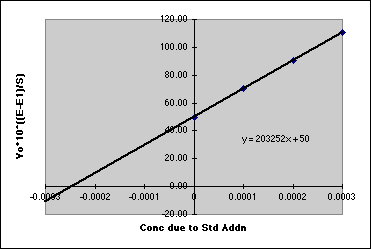
Some classic errors that students make on this report are (1) Totally forgetting to do the standard addition. Once they complete the stuff on the textbook method, they feel they are done. (2) When doing the standard addition, they simply add their standards to their unknown, usually producing a massive increase in concentration that wipes out any response from the unknown itself. Adding .01 M F- to a 0.0001 M F- unknown pushes the intercept down into the noise. You must think through how you will do the standard addition. The plots shown above for fluoride will be totally inappropriate for gas chromatography. This may take some effort.
Remember, the discussion of what your data means is the largest part of your report. You must make the case that your method is correct and reliable within the error limits that you compute. To say simply that it seemed to work is not enough. Although Standard Addition cannot remove all possible Matrix effects, comparison of the Working Curve method with Standard Addition is your best tool for evaluation of the method that you have on this report. Don't forget to be quantitative about the errors involved. Use the values you have generated. Don't simply characterize results as "good" or "bad."
Grading
Each experiment with a quantitative unknown is worth 100 points. In addition there will be 20 points on the first two experiments for notebook organization. Reports and other laboratory work will add about 200 points more to your lab score. Laboratory counts for 40% of your course grade. You must pass both the laboratory and the lecture to pass the course. Passing in the laboratory is generally set at 60%. See the laboratory schedule sheet for due dates and penalties.