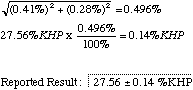
The Lab Notebook
--Basic Format
--Data Summary and Expected Statistics
Laboratory Procedures and Waste Management
--Chemical Waste
--Care of the Balances and Weighing Procedures
--Table of Available Reagents (Memorize Concentrations)
--Drying and Storing Solid Chemicals
--Cleaning Glassware
--Gravimetric Analysis of Chloride
--Glassware calibration
--Volumetric Chloride
--The KHP Experiment and Titration Curve
--Determination of %CaO in a Solid Unknown by EDTA Titration
--Spectrophotometric Iron
--Determination of Iron in an Ore by titration with Potassium Permanganate
--Determination of Glucose
You must use a bound laboratory notebook. Your instructor will show you an example. Reserve the first four pages for a table of contents, then number all succeeding pages. Leave the left-hand page blank for scratch. Put all hard data and calculations on the right hand side. All entries must be in ink. Nothing can be erased; pages must not be removed. If you make an error, cross it out and leave a note explaining why the entry was voided. It is very important that ALL INFORMATION be placed DIRECTLY into your notebook. Do not use paper towels or other intermediate pieces of paper to record data for later transferral into your notebook. There is plenty of room in your notebook for all of your experiments, so leave ample space for data tables and notes. The important thing is to have all of your data clearly labeled and reasonably well organized. A little planning will make this possible. You will be downgraded for unreadable and unlabeled entries. Also note that a Data Summary is called for at the end of each experiment; here is where you can indulge your urge to exclude unusable data and produce neat tables.
Each experiment should follow this format:
1. Use a clear Title to begin each new experiment. In Chem 105 we ask that each experiment be given its own section of your notebook. When starting the preliminary work on the next experiment, leave enough pages in between so the preceding experiment can be finished up without overlap. (Some commercial laboratories may require you to keep a daily log of activities in your notebook, which may result in many experiments appearing on the same pages. Under these conditions, proper labeling of each activity becomes even more critical.)
2. Provide a brief theoretical discussion of the experiment involved. Include all relevant chemical reactions and mathematical procedures. Do this before you start the experiment. The purpose is to make you organize all of the theoretical details so they are clear in your mind.
3. Make an outline of the experimental procedures used in the experiment. Only rarely will we be doing the experiment exactly as specified in the book or on a handout. This outline, done before you start the experiment, will allow you to think through the procedures, and to collect all of the relevant details into one place. Do not simply copy the sections of your text or handout into your notebook, but condense the procedures into an easy-to-follow guide. References to lengthy descriptions should replace the descriptions themselves. (e.g. "Dissolve the calcium carbonate by adding HCl to an aqueous suspension and heating. See the handout titled 'CaO by EDTA' for details.")
4. Place all measurements into Data Tables. Make these as clear and as neat as possible. Be sure all data are properly labeled so you can find them later. Carry your notebook to the balance or other instrument for direct entry of all data. If you find things get a little messy at his point, don't worry too much. Just make sure you can identify all of the numbers two weeks later. You will be graded on clarity, not neatness. Remember the data summary at the end of the experiment will let you present your data in a highly organized way, while preserving your original entries.
5. Gather together all of the data required to do your calculations into a Data Summary. For example, the results from a series of Standardization titrations might appear as:
Grams KHP
|
mL
NaOH used
|
Molarity
|
Comments
|
| 0.5103
g
|
23.78
mL
|
overshot
e.p.
| |
| 0.6236
g
|
28.46
mL
|
0.1073
M
|
|
| 0.6022
g
|
27.33
mL
|
0.1079
M
|
|
| 0.5578
g
|
25.41
mL
|
0.1075
M
|
|
| 0.5971
g
|
27.12
mL
|
0.1078
M
|
Sample Calculation:
0.5103 g KHP
|
mole
KHP
|
1
mole NaOH
|
|
| 204.23
grams
|
1
mole KHP
|
0.02378
L NaOH
|
Mean Molarity 0.1076 M
Standard Deviation 0.000275 M
95% Confidence limit 0.000438 M
Relative CL (%) = (0.000438/0.1076) x 100% = 0.41 %
With your data all in one place, performing the calculations will be much easier. If you make a calculation error resulting in garbage for results, finding this error will be much easier. If data have been miscopied from your original entries, the correction can easily be made by comparing the earlier entries in your notebook.
6. Compute your results and place them with your data, perhaps in the same table as shown above (grams and milliliters are data; molarities are results). Always include at least one complete sample calculation so that all constants and equations can be checked.
If you have used a computer to enter your data and calculate your results, perhaps by using a spreadsheet program, print out your tables and graphs and paste them into your laboratory notebook. Remember to include only the results that are needed for your experiment. Computers can generate a lot of output very quickly. Resist the temptation to paste in everything you have created, just because you think it looks impressive.
7. Do a statistical analysis of the results. A sample is shown in the example above. All experiments will require 95% confidence limits and other statistics. Reminder: this should not be the first time you have computed the errors as shown above. Such statistics must be computed as soon as the data are collected. If your relative errors are much larger than 0.2% - 0.5% (for most titration experiments), there is something wrong with your technique. See your instructor for help in correcting your laboratory methods before any more data are collected. The time to discover poor results is during the experiment, when there is time to make corrections. You do not want to discover this while making your final data and results summary!
To the example give above, you would have to add a second section for your unknown runs. In computing the final error for your unknown, don't forget to combine the errors from the unknown and the standardization runs to produce the overall error for the experiment. e.g. suppose your unknown runs result in a mean of 27.56 %KHP with a relative CL of 0.28%. (Note the difference between % and %KHP). The combined relative CL will be
8. You experiment concludes with a final reported result, as shown above. (In Chem 105 we generally report our results to the four significant figures we have used when recording milliliters and grams. In the real world the result shown would have to be rounded down some more and reported as 27.6 +/- 0.1 %KHP.) Make any comments you wish about the quality of your results, and include suggestions for improving the experiment.
Note that you will never surrender your notebook to your instructor for grading. We will go over it together at the time you report your results. Grades will be awarded on the spot.
You are required to wear SAFETY GLASSES in the laboratory at all times. Purchase a pair that is comfortable and has good optical qualities. Although goggles offer the best protection, students find that they steam up and are uncomfortable. The best compromise is probably the the more comfortable type that looks like regular glasses with side shields. Contact lenses are NOT allowed in the laboratory. They are hard to remove in case of an accident; soft lenses also absorb laboratory fumes and rapidly become cloudy and irritating to your eyes.
You will need a good calculator with scientific and statistical functions. Remember to calculate your statistics as soon as you collect your data, so that problems can be found and corrected while there is still time.
To use some of the computers in the Macintosh computer lab on the second floor of Natural Science you will need a Macintosh Start-Up Disk. (Half of the ten computers will not need this disk and will boot up from an internal hard drive.) Purchase this disk at the local Bookstore, as well as some blank 3.5" double sided double density floppies for storing your data. Warning! Eight of the Macs in S-232 are very old and can only use 800K 2S2D floppies. Do not attempt to use 1.44MB HD floppies with these old machines. Although it may appear that your HD disk formats on these old machines to 800K, the data generally fades away in about two weeks, since the old drives can't reliably magnetize the HD disks. Although you can do graphing and least squares analysis without a computer, it is time to start using things like spreadsheet programs and word processors routinely. If you know nothing about using computers, your instructor will give you enough information to get started.
Camera stores and cosmetics counters sell white cotton gloves. They work well for handling materials in the balance room. Purchase of these is optional, but you will find them a great convenience.
Laboratory Procedures and Waste Management
There are certain things you must do in lab to protect yourself, your data, and the outside world (pollution). The following list is not exhaustive, but does cover behavior often seen in real laboratories.
1. Check with your instructor before discarding chemicals down the sink. Waste containers are provided for heavy metals, certain organics, and other environmental nasties. If you see the waste container getting full, notify your instructor.
2. Take care of your analytical balance. Each time you leave your balance make sure that the balance is clean, the doors are closed, the pan arrest is set (mechanical balance), and the balance weights are set back to zero (mechanical balance). Before you leave the lab, be sure the cover is placed back over the balance. When using your balance, remove the plastic cover completely. If the cover is only pushed back on a mechanical balance, heat from the lamps cannot escape, which may cause problems. It is recommended that you run the calibration on electronic balances each day before first use.
3. Make no messes in the balance room. Virtually all weighings in your analytical balance are made by difference in closed weighing bottles, so there is no(?) chance of chemical spillage. If you happen to spill some anyway, CLEAN IT UP IMMEDIATELY! (Your instructor will illustrate use of the balance, and how to Weigh By Difference.)
When weighing larger quantities of chemical on the top-loader, be sure to use a small beaker or watch glass to hold the chemical (use the tare adjust to zero out your container). If you must use weighing paper, fold it into a little box first. Again, if you spill something, CLEAN IT UP IMMEDIATELY.
Small bottles of reagent solids are often available in the balance room. Replace the caps of these containers immediately after use. NEVER return excess chemical to a reagent bottle. Bring a waste container with you if you think you will need one.
Do not waste time trying to duplicate masses called for exactly. If the experiment tells you to weigh out "0.2 g" do not spend hours in the balance room trying to get 0.2000 g! The 0.2 g is called a TARGET value. Try to weigh out to within +/-10% of this value, but do not waste time trying to get it dead on. Record the value you did weigh in your laboratory notebook.
4. When you are issued an unknown solid, you generally transfer most of it to a weighing bottle. Label the bottle, keep the bag, and write your unknown number in your laboratory notebook. (In general you will be using the same unknown number all semester, but certain repeats will be done with a different number.) Remember, if you lose your unknown somehow, it will cost you 20 points to get more.
5. Make no messes in the hoods. Our concentrated reagents are stored there. Bring a small container and transfer only the amounts you think you will need for immediate use. If you spill something, CLEAN IT UP.
Note that the concentrations of these reagents are not stated on the labels. Here is a table of concentrations that you MUST MEMORIZE (well... at least know hydrochloric, nitric, and sulfuric acids, and ammonia).
Name
|
Approximate Molarity
|
| Acids:
|
|
| Acetic
|
17.4
M
|
| Hydrochloric
|
12.1
M
|
| Hydrofluoric
|
28.9
M
|
| Nitric
|
15.9
M
|
| Perchloric
|
11.7
M
|
| Phosphoric
|
14.8
M
|
| Sulfuric
|
18.0
M
|
| Bases:
|
|
| Ammonia
|
14.5
M
|
| Sodium
Hydroxide (50% w/w)
|
19.4
M
|
| Potassium
Hydroxide (45% w/w)
|
11.7
M
|
6. When using the ovens, always check the temperature first. If something seems amiss, inform your instructor. Do NOT attempt to correct oven temperatures yourself. When drying chemicals in an oven, the materials are generally placed in an open weighing bottle, which is set in its cover, which is placed in a beaker, which is covered with a ribbed watch glass. Label the beaker and the weighing bottle by writing in pencil on the frosted areas provided. Do NOT use paper labels.
Open oven doors for as short a time as possible to maintain the internal temperature. At times your instructor will designate certain ovens for special uses. Be sure you are using the correct oven.
Store dried chemicals in weighing bottles kept in your desiccator.
7. Glassware is generally cleaned with hot soap and water, followed by a rinse with tap water, and a final rinse with distilled water. Use a clean towel to wipe the glassware dry. We rarely need to dry glassware in an oven, since we will soon be getting it wet again anyway.
Burets will need to be cleaned with special cleaning solution before rinsing with distilled water. Remember that any drops of reagent left clinging to the inside of your buret produces instant error. (Other common sources of buret error are failing to rinse the buret with a little reagent before filling, failing to split the drops at the end point, failing to reproduce the end-point color, and failing to record volumes to +/-0.01 mL.)
8. Be prepared when you come to lab. Before coming to lab, read the materials provided as well as your notes, and prepare the preliminary portions of your notebook as described above. Do not just show up and ask someone what to do.
1. Check in and balance instruction -- this will be covered in the lab. Your introduction to the balance and many of the concepts involved should be considered part of the lecture material and may appear on a lecture exam.
2. Gravimetric Analysis of Chloride -- You will be issued enough Chloride unknown to do both this experiment and the next one on Volumetric Analysis. You will also be given a bag of silver nitrate that is used to prepare 500 ml of 0.1 M AgNO3 -- this too must last through both experiments.
Clean and dry all of your weighing bottles. Put an identifying label on each bottle and its cover(use pencil on the frosted areas provided), then weigh each bottle with its cover in place. and record this TARE weight in your laboratory notebook. Transfer your chloride unknown to one or more weighing bottles (never fill over 2/3). Do the same with the silver nitrate. Place them in covered beakers (use a ribbed watch glass) and dry both materials for one hour at 110deg.C. After they have cooled in your desiccator, weigh the bottle again to see how much material you have been given.
After the unknown has remained in the desiccator overnight, samples may be weighed by difference. Your instructor will illustrate this technique.
Weigh out enough silver nitrate to make up the 500 mL of 0.1 M solution. Remember, the weight need be only within 10% of the nominal, or TARGET value required. We will use this silver nitrate solution in place of the one called for in the book for gravimetric analysis.
Clean and dry three medium (best) or fine (OK, but filters slowly) porosity sintered glass crucibles. (If you have a coarse porosity crucible, return it to the storeroom and get a medium to replace it.) Bring these crucibles to constant weight with repeated dryings.
Additional procedures are in the book or will be discussed in lab.
3. Glassware calibration -- although a day has been put into the schedule for this, it is best done while drying the crucibles in the Gravimetric Chloride experiment.
4. Volumetric Chloride -- This is done on the same unknown sample used for the gravimetric analysis. You will also be issued some standard sodium chloride for standardizing the silver nitrate.
Most recent textbooks have eliminated the "Mohr Method" in favor of an adsorption indicator method that does not add chromium salts to the laboratory waste stream. Your instructor may choose either the adsorption method or the Mohr Method for your experiment. (The Mohr Method is a classic example of a differential precipitation.) See the laboratory schedule. The procedure for the adsorption method will be in your textbook.
5. The KHP Experiment is done as indicated the lab schedule, but also includes a titration curve. The following instructions are for the titration curve part only.
The titration curve is to be run on three millimoles of standard KHP. Do not run the curve on your unknown. Consider the titration curve to be an independent experiment.
A. Setting up for the Titration Curve
You will need a pH meter, a combination pH electrode, a stirring bar, a magnetic stirrer, your buret, and the smallest beaker that will hold the electrode, the stirring bar, and the tip of the buret. Position the beaker on the stirrer so that the stirring bar can rotate slowly and smoothly. It should not jump up, splashing liquid out of the beaker and smashing a $100 pH electrode. The pH electrodes are generally kept in a pH 7 buffer solution between uses. Be sure to remove the electrode from its storage container and rinse it off before use.
Your instructor will show you how to plug the electrodes into the pH meter. If you have a "Beckman-Style" connector, be sure that the large pH electrode jack is pushed firmly into its receptacle and tightened down against the spring pressure. This will take two hands.
Calibrate your pH meter by first using a pH 7.00 buffer and adjusting the CALIBRATE know to read 7.00 on the display. then rinse your electrode, go to a pH 4.00 or 10.00 buffer and use the SLOPE knob to set the buffer pH. Remember to push the OPERATE button while taking readings, and the STANDBY button when changing solutions or handling the electrodes. It is possible to damage a pH meter with a static electricity shock by handling electrodes in the OPERATE mode.
B. Solutions
Your buret holds the 0.1 M NaOH. The beaker holds three millimoles of KHP and enough distilled water to cover the active portions of the combination electrode. Add enough Methyl Red to give a distinct red color; add the usual amount of phenophthalien -- usually three drops or so. Be sure that the electrodes are high enough to keep the fragile electrode bottom away from the stirring bar!
C. Running the Curve
Set up a full page in your notebook for data. Use column headings of Milliliters, pH, and Color Changes. On the facing page set up a graph for a quick plot of your results as you get them. The ordinate should be from 0 to 14 pH; the abscissa should be 0 to 50 ml.
Take pH readings every 2 ml or so until you get close to the end point region. Your quick plot will show you the upturn just before the end point region. Now proceed dropwise, using microsquirts in the steepest central part of the curve, recording ml, pH and color changes every 0.1 to 0.2 change in pH. Continue until the complete S-shape of the curve is obtained. The most common errors made here are not to plot the data, go too quickly through the endpoint region, and leave large gaps in the curve. You will be downchecked if this happens.
D. The Titration Curve Report
Your report will be turned in on separate paper, not in your notebook. It consists of the following:
A table of your data (You may turn in a Xerox of your notebook if it was done neatly, and you elected not to use a computer on your data.)
A plot of the entire titration curve, with color changes indicated on the graph. The plot must be done carefully on good graph paper if you elect to plot this by hand.
Students are encouraged to use a computer spreadsheet program with graphics to do the graphs for this experiment. Although formal computer training is not part of this class, your instructor will be happy to show you how to do this on the Macintosh computers available in New Science. It should take less than an hour to learn how to do this, even if you have never touched a computer before. You will need a Macintosh StartUp Disk (bookstore) and a blank data disk to use some of the computers.
An expanded graph of the end point region with color changes, possible carbonate error, instrumental end point (steepest part of the curve), color change end point, and titration error indicated.
Calculate the estimated titration uncertainty in milliliters and in relative % for both color changes and for the instrumental end point. Remember, there is no such thing as a perfect measurement. You have to estimate the width (in milliliters) of the "fuzzy" area within which your end point could have fallen. There is no direct way to calculate this. Look for the minimum and maximum possible volumes that begin and end an end point region. You have three end points to consider: methyl red, phenolphthalien, and instrumental (based on the shape of the titration curve). Use a table to present your results. It is the purpose of this part of the report to sharpen your professional skills in error estimation. ["How well can you measure that?" "Oh, within about 1% on a good day; it isn't all that good."]
Make a Critical Conclusion. Summarize your calculations of error, comment of the sizes of the errors and the suitability of each end point method for analysis. Discuss why you think carbonate error may, or may not, be present. Remember to be quantitative in your discussion of errors. Don't just call them "big" and "little" or "good" and "bad." Use the data you spent so much time collecting.
6. Determination of %CaO in a Solid Unknown by EDTA Titration
The procedures in your text are probably designed for aqueous samples containing calcium and magnesium. (See the references in your lab schedule to find the procedure for "hard water" analysis.) To use solid unknowns and standard materials, you will need to dissolve them first. Since calcium carbonate will not dissolve in distilled water, you will need to follow the directions below before completing the titrations as directed in your text. We have found that the purity of our EDTA reagents is not good enough to use it as a primary standard. Directions for preparing a standard calcium solution are also given. Titrate this solution in the same way you titrate your unknown solution; then calculate the molarity of the EDTA.
A. Your instructor will provide you with bags of unknown, of primary standard calcium carbonate, and of the EDTA. Dry the unknown and the calcium carbonate for one hour at 110deg. C. The EDTA should not be dried; just put it in a weighing bottle.
B. Prepare 500 ml of 0.01M EDTA. The salt is usually Na2H2Y.2H2O. (Y = EDTA) Compute the required mass, weigh it out, and make up to the mark with distilled water in a 500 ml volumetric flask. The exact molarity of this solution must be determined by standardizing against standard calcium carbonate.

Because of the subtleties of the red to blue color changes, it is recommended that you NOT wear bright red on EDTA days.
C. Many EDTA procedures require that traces of magnesium be added to your solution. To avoid adding any additional titratable cation (Mg2+) we add the magnesium as MgY2-. To prepare this complex (hereafter referred to as "the spike"), weigh out 0.4 millimoles of Mg2+. Check the reagent bottle for composition and gram formula weight needed to calculate the mass. The salt is usually MgSO4, but could be something else. Dissolve in 50 ml of distilled water, and titrate according to the hard water instructions in your text. In order to experiment with the end point, you can add a crystal of Mg salt to restore the red color, then run through the end point again. Stop at your best estimate of the end point color. Store the solution in your glass stoppered erlenmeyer flask. Label it "Mg Spike Solution." When titrating either standard calcium, or a calcium unknown, add about one ml of this spike to your solution in addition to the usual buffer and indicator. (Note: Your text may have an alternate recipe for "spike" solution. Follow the one in these notes, not the one in your text.)
D. Dissolution of solids containing calcium carbonate requires special procedures--this includes both your standard calcium carbonate as well as your unknown. Once the solid is weighed out (see below) transfer it to a 250 ml erlenmeyer flask, and add about 100 ml of water. Add 6M HCl dropwise and swirl the flask to dissolve the calcium carbonate. Warm after each addition to expel the carbon dioxide. Continue until all of the solid has dissolved. (In the case of many unknowns, a small amount of insoluble white silica may remain.) Avoid a large excess of the HCl. After dissolution is complete, quantitatively transfer to a suitable volumetric flask and dilute to the mark. Shake for five to ten minutes to achieve uniformity.
E. To prepare a standard solution, weigh enough dried calcium carbonate standard to allow you to use 25 ml aliquots from a 250 ml volumetric flask. (The calculation of the mass is left as an exercise for the student. Remember that each titration will require at least 0.3 millimoles of calcium.) After preparing the solution and mixing it well, transfer to another container to free the volumetric flask for your unknown.
F. The unknown solution is prepared by dissolving about one gram of unknown (see step 4) and making up to 250 ml in a volumetric flask.
G. The percentage of CaO in the unknowns can vary considerably from student to student. Try a 10 ml aliquot first. If the titration volume is too small, choose an appropriate larger aliquot size.
H. Don't forget to report %CaO. This is the standard way to report rock and mineral analyses. It does not depend on the actual compound of calcium in the sample.
Note: Buffer and indicator solutions are available in the lab. Be sure you use the high buffer capacity pH 10 solution designed for this experiment. Do not confuse it with the low buffer capacity pH 10 solution used to calibrate pH meters. pH paper will be available to check the pH of your solutions if your end point seems too drawn out due to low pH. The indicator will be either Calmagite or Eriochrome Black T. See your instructor.
On the surface this experiment is a very simple one. If you add sodium acetate to buffer a solution containing iron, reduce the iron to iron (II) with hydroxylamine hydrochloride, and then add 1, 10 - phenanthroline to form an orange complex, you can measure the amount of Fe present using a spectrophotometer and Beer's Law (A = abC). Specific procedures for doing this are found in your text.
The tricky part is preparing suitable standard iron solutions for calibrating the spectrophotometer. The absorbances of the solutions should lie between about 0.1 to 0.8 absorbance. A concentration of about 1 microgram Fe/mL should fall within this range. It will be up to you to devise a way to prepare accurately four standard solutions that will cover this range.
Here is what you have to work with. A bag of ferrous ammonium sulfate will be used as a reasonably good primary standard. Place this in a weighing bottle and keep it in your locker. Do not dry or desiccate ferrous ammonium sulfate--you might change its water of hydration. To measure volumes you have your buret, your class-A pipets and your volumetric flasks. The 50 ml vol flasks should be used for most of your calibrating orange solutions. Larger vol flasks are used for dissolved unknown and standard stock solutions. Remember that volumes less than 5 ml cannot be measured accurately with this equipment.
The usual procedure is to make up 500 to 1000 ml of ferrous ammonium sulfate stock solution. This is then diluted to make an intermediate Fe stock of 100 to 250 ml. Portions of the intermediate stock are added to the 50 ml vol flasks, color forming reagents are added, and the orange color developed. Measurement of the absorbances of these standard solutions are used to make a standard Working Curve of Absorbance vs. Concentration.
You will also be issued a solid Unknown. Dissolve this with HCl according to the directions given for the Redox titration of an iron ore, and dilute to 500 or 1000 ml to make an unknown stock. Dilute this stock appropriately until it can be added to a 50 ml vol flask to which the color forming reagents will be added. Measure the absorbance and calculate the concentration from your standard working curve.
You are not finished yet. Although you now have a value for your unknown iron concentration, the number is fairly crude. Prepare a new unknown iron dilution that brings its value near to the ideal 0.3 absorbance that gives minimum instrumental error. Prepare a new series of standard solutions that fall closely around the value of the unknown iron. Run the unknown and the standards all at the same time to minimize error. Repeat the determination a few times to give you some statistics to work with.
This experiment requires a fair amount of planning on your part. Your instructor will give you more information in class.
Atomic Absorption (AA): Once you have your dilution scheme worked out for the Spectronic 20, you should be able to use the same solutions to generate a working curve and determine your unknows using Atomic Absorption. Your lab instructor will demonstrate how to use the AA instrument. After running your standards and samples, evaluate how well the results work. If necessary, change the dilutions to get better results by AA. (AA usually requires higher concentrations, perhaps five or ten times more concentrated than for the Spectronic 20. Do a sample run to find out for sure.)
Your grade will be based on 50 points for the Spectronic 20 result, and 50 points for the AA result. The intrinsic error for both instruments is roughly the same, so one result should not influence the other.
8. Determination of Iron in an Ore by titration with Potassium Permanganate.
The procedure in your text includes directions for preparing the permanganate and standardizing it against sodium oxalate.
Determine the same unknown used in the spectro iron experiment. The procedure in the book should work as stated. The tin chloride, mercuric chloride, and Zimmermann-Reinhardt reagents will all be available in the lab. Remember you will not be allowed to begin this experiment until after you have reported the results from spectrophotometry.
9. Determination of Glucose -- See the procedure below. Make sure you wade through all of the chemistry and have a good outline in your laboratory notebook. It is easy to mess up this experiment if you don't know what you are doing.
Reducing sugar (glucose for example) determinations are routinely run on samples of blood, urine, various beverages and food products. A general method for determining reducing sugar follows.
It is common knowledge that Fehling's solution (which contains Cu+2) is reduced by glucose to form a brick-red precipitate of Cu2O. The amount of Cu2O formed may be determined by oxidizing it back to Cu+2, using free iodine. The iodine used for this purpose is generated in the solution by the reaction of KI with KIO3 in the presence of acid:
5 I- + IO3- + 6 H+ ---> 3 I2 + 3 H2O (1)
Oxidation of the Cu2O precipitate may be represented by equation (2)
Cu2O + I2 + 2 H+ ---> 2 I- + 2 Cu2+ + H2O (2)
Equation (1) forms a known quantity of excess iodine. After reaction (2) is complete, the remaining iodine (which did not react with the Cu2O) is determined by tritation with standard sodium thiosulfate solution using starch indicator:
2 S2O32- + I2 ---> 2 I- + S4O62- (3)
The overall reaction that defines the equivalent weight of KIO3 is the following:
IO3- + 5I- + 6H+ + 6S2O32- ---> 6I- + 3S4O62- (4)
Where iodate to iodide is a six electron change.
Solution A: Dissolve 34.64 g CuSO4.5H2O in water and dilute to 500 ml.
Solution B: Dissolve 173 g sodium potassium tartrate and 50 g NaOH in water and dilute to 500 ml. (A and B will be available in the laboratory.)
Standard potassium iodate solution: Weigh to the nearest 0.1 mg enough dry KIO3 (110o, 1 hr) to prepare 500 ml of about 0.1 N (1/60 M) solution. Quantitatively transfer the salt to a 500 ml volumetric flask and dissolve in a small amount of water. After first making the solution basic with 3-4 drops of 6 N (6 M) NaOH add about 20 g of iodate-free KI. Dilute to 500 ml.
Sodium thiosulfate solution: 0.12 N (0.12 M). Dissolve 19 g of Na2S2O3 (or enough Na2S2O3.5H2O to give 19 g of Na2S2O3) in 1 liter of distilled water. Add about 0.1 g of Na2CO3 to keep the solution neutral or slightly alkaline and thereby retard decomposition to elemental sulfur. If you wish, you may prepare this solution quantitatively as a check on the normalities (molarities). See G.D. Christian, 3rd Ed., p. 278, for additional precautions regarding thiosulfate solutions.
Starch indicator (Make daily): Suspend 1 g of soluable starch in 100 ml distilled water. Bring to a boil. Cool and use 1-2 ml per titration.
Saturated potassium oxalate: Available in laboratory.
Dry the sugar sample for not more than 1 hour at 95oC. Weigh about 2 grams of the sample to +/- 0.1 mg and place it in a 500 ml volumetric flask. Dissolve in about 300 ml of water and then fill to the mark. Mix thoroughly. Prepare a boiling water bath of 1 liter of distilled water in a 2-liter beaker. Rapidly pipette 25 ml each of Fehling's solutions A and B into a 500 ml Erlenmeyer flask. Complete the following analysis before doing additional runs. Add exactly 50 ml of the glucose solution prepared above. (Note: If a sample smaller than 50 ml is to be analyzed, the total volume should be brought to a100 ml at this point by addition of water). Cover the flask with a small inverted beaker and place in the boiling water bath for exactly 10 minutes. (Note: The bath must be large enought to boil the entire 10 mintues.) After 10 minutes immediately cool the flask to room temperature with running water or ice bath.
Add 20 ml of saturated potassium oxalate solution and mix. This prevents future oxidation of iodide by copper (II) ions and dissolves any Cu2I2 formed. Pipette 50 ml of KIO3 solution into the flask followed by 16 ml of 6 N (3 M) H2SO4 (measure with a graduate). Shake the solution gently until the copper(I) oxide is completely dissolved. If insufficient iodine is evolved add another portion of KIO3 solution or start with a smaller aliquot of sugar sample.
Titrate the remaining I2 (actually brown I3-) with sodium thiosulfate solution, using starch indicator. The starch should be added just before the endpoint is reached, as indicated by the color of the solution. (Why?)
Standardize the sodium thiosulfate solution by repeating the above procedure, omitting the Fehling's solutions and potassium oxalate, substituting 120 ml of distilled water for the sugar sample and using 25 ml of KIO3 solution instead of 50 ml. Calculate the normality (molarity) of the sodium thiosulfate solution. Calculate the copper equivilent for your unknown as mg of copper in the Cu2O. Convert mg Cu to mg glucose using one of the methods suggested below and report your answer as mg glucose/100 ml based on an unknown sugar sample weighing exactly 2.000 grams.
Methods for converting mg Cu to mg glucose:
(1) If a chemistry handbood or other source of Munson-Walker- Hammond tables is available, read mg glucose equivilent to your mg Cu directly from the table.
(2) From an accurately plotted graph or an equation based on the following Munson-Walker data as determined by Hammond, determine the Cu-glucose weight ratio.
Cu in mg Cu-glucose ratio400 1.863 350 1.892 300 1.921 250 1.950 200 1.978 150 2.008 100 2.041 70 2.059 50 2.075
Divide the amount of copper in your sample by the Cu-glucose ratio.
Both of these methods give glucose in mg/50 ml if a 50 ml sugar sample was used. Please remember that you are to report your answer as mg glucose/100 ml, based on a glucose sample of exactly 2.000 grams. Correct your result for the actual mass of sample used.
A worked out example of this calculation is found here. If you have a JavaScript-capable browser (Netscape 2.0 or better) you can check your copper/glucose ratio calculation with the calculator found there.